Поскольку в кишечнике всасываются все поступающие с пищей моносахариды (фруктоза, галактоза, манноза и т.п.), то перед организмом встает задача превратить полученные гексозы в глюкозу для ее дальнейшего использования в реакциях метаболизма – происходит превращение сахаров. При дефекте соответствующих ферментов возникает накопление моносахаридов в крови – галактоземия и фруктоземия.
Превращение моносахаров
Цель этого процеса – создание только одного субстрата для реакций метаболизма, а именно α-D-глюкозы, что позволяет сэкономить ресурсы, не образовывать множество ферментов для каждого вида моносахарида. Реакции образования свободной глюкозы протекают в эпителии кишечника и, в основном, в гепатоцитах.
Превращение галактозы
Галактоза сначала подвергается фосфорилированию по 1-му атому углерода. Отличительной особенностью является превращение в глюкозу не напрямую, а через синтез УДФ-галактозы из галактозо-1-фосфата. Источником УМФ является УДФ-глюкоза, имеющаяся в клетке. Образованная УДФ-галактоза впоследствии изомеризуется в УДФ-глюкозу и далее ее судьба различна.
Она может:
- участвовать в реакции переноса УМФ на галактозо-1-фосфат,
- превращаться в свободную глюкозу и выходить в кровь,
- отправляться на синтез гликогена.
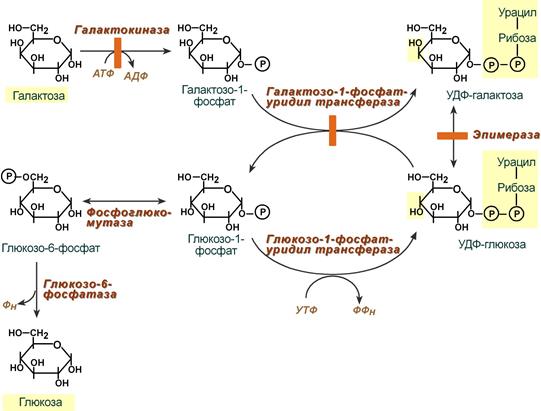
Превращение галактозы в глюкозу
(обратимость обеих уридил-трансферазных реакций не показана)
Биохимическое усложнение вроде бы простой реакции эпимеризации вызвано, видимо, синтезом УДФ-галактозы из глюкозы в молочной железе для получения лактозы при образовании молока. Также галактоза используется при синтезе соответствующих гексозаминов в гетерополисахаридах.
Нарушения превращения галактозы
Нарушения обмена галактозы могут быть вызваны генетическим дефектом одного из ферментов:
- галактокиназы, частота дефекта 1:500000,
- галактозо-1-фосфат-уридилтрансферазы, частота дефекта 1:40000,
- эпимеразы, частота дефекта менее 1:1000000.
Заболевание, возникающее при этих нарушениях, получило название галактоземия.
Диагностика. Дети отказываются от еды. Концентрация галактозы в крови возрастает до 11,1-16,6 ммоль/л (норма 0,3-0,5 ммоль/л), в крови появляется галактозо-1-фосфат. К лабораторным критериям относятся также билирубинемия, галактозурия, протеинурия, гипераминоацидурия, накопление гликозилированного гемоглобина.
Патогенез. Избыток галактозы превращается в спирт галактитол (дульцитол), накапливающийся в хрусталике и осмотически привлекающий сюда воду. Изменяется солевой состав, нарушается конформация белков хрусталика, что приводит к катаракте в молодом возрасте. Катаракта возможна даже у плодов матерей с галактоземией, употреблявших молоко во время беременности.
При дефекте галактозо-1-фосфат-уридил-трансферазы АТФ постоянно расходуется на фосфорилирование галактозы и дефицит энергии угнетает активность многих ферментов, "токсически" действуя на нейроны, гепатоциты, нефроциты. Как результат возможны задержка психомоторного развития, умственная отсталость, некроз гепатоцитов и цирроз печени. В почках и кишечнике избыток галактозы и ее метаболитов ингибирует всасывание аминокислот.
Основы лечения. Исключение из рациона молока и других источников галактозы позволяет предотвратить развитие патологических симптомов. Однако сохранность интеллекта может быть достигнута только при ранней, не позднее первых 2 месяцев жизни, диагностике и вовремя начатом лечении.
Превращение фруктозы
В целом переход фруктозы в глюкозу осуществляется по двум направлениям. Сначала происходит активация фруктозы посредством фосфорилирования либо 6-го атома углерода при участии гексокиназы, либо 1-го атома при участии фруктокиназы.
В печени имеются оба фермента, однако гексокиназа имеет гораздо более низкое сродство к фруктозе и этот путь превращения слабо выражен. Образованный ею фруктозо-6-фосфат далее изомеризуется и глюкозо-6-фосфатаза отщепляет уже ненужный фосфат с получением глюкозы.
Если работает фруктокиназа, то образуется фруктозо-1-фосфат, под действием соответствующей альдолазы он превращается в глицеральдегид и диоксиацетонфосфат. Глицеральдегид фосфорилируется до глицеральдегидфосфата и вместе с диоксиацетонфосфатом они в дальнейших реакциях либо используются в гликолизе, либо в реакциях глюконеогенеза превращаются в фруктозо-6-фосфат и далее в глюкозу.
Особенностью мышц является отсутствие фруктокиназы, поэтому фруктоза в них превращается сразу в фруктозо-6-фосфат и поступает в реакции гликолиза или синтеза гликогена.
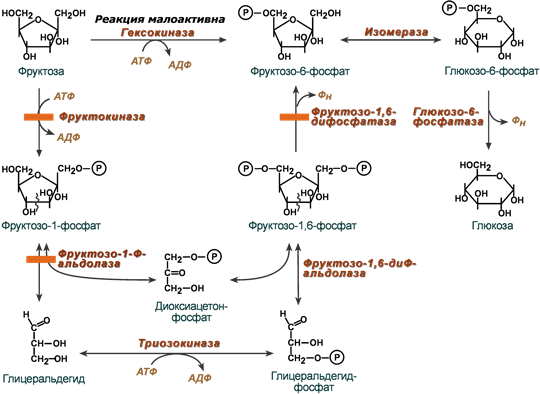
Пути метаболизма фруктозы и ее превращение в глюкозу
Нарушения метаболизма фруктозы
Эссенциальная фруктозурия
Генетический дефект фруктокиназы приводит к доброкачественной эссенциальной фруктозурии, протекающей безо всяких отрицательных симптомов.
Наследственная фруктозурия
Заболевание формируется вследствие наследственных аутосомно-рецессивных дефектов других ферментов обмена фруктозы. Частота 1:20000.
Дефект фруктозо-1-фосфатальдолазы, которая в норме присутствует в печени, кишечнике и корковом веществе почек, проявляется после введения в рацион младенца соков и фруктов, содержащих фруктозу.
Патогенез связан со снижением мобилизации гликогена из-за ингибирования гликогенфосфорилазы фруктозо-1-фосфатом и ослаблением глюконеогенеза, т.к. дефектный фермент способен участвовать в реакциях аналогично фруктозо-1,6-дифосфат-альдолазе. Проявляется заболевание снижением концентрации фосфатов в крови, гиперфруктоземией, тяжелой гипогликемией. Отмечается вялость, нарушения сознания, почечный канальцевый ацидоз.
Диагноз ставится исходя из "непонятного" заболевания печени, гипофосфатемии, гиперурикемии, гипогликемии и фруктозурии. Для подтверждения проводят тест толерантности к фруктозе. Лечение включает диету с ограничением сладостей, фруктов, овощей.
Дефект фруктозо-1,6-дифосфатазы проявляется сходно с предыдущим, но не так тяжело.