Гормональная регуляция
Синтез и окисление триацилглицеролов и жирных кислот зависит от соотношения инсулин / глюкагон.
1. Изменение количества ферментов
Ферменты комплекса пальмитатсинтазы и ацетил-SКоА-карбоксилазы являются адаптивными ферментами, количество их возрастает при усиленном питании и уменьшается при голодании и потреблении жира. Индуктором биосинтеза этих ферментов является инсулин.
2. Ковалентная модификация
Благодаря инсулину, глюкагону, адреналину, тиреотропному и адренокортикотропному гормонам происходит ковалентная модификация ферментов ацетил-SКоА-карбоксилазы и ТАГ-липазы путем фосфорилирования-дефосфорилирования.
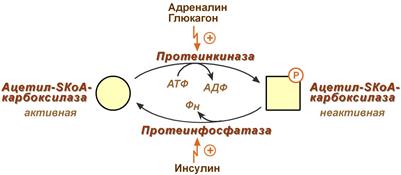
Регуляция активности ацетил-SКоА-карбоксилазы
Инсулин активирует протеинфосфатазу и способствует дефосфорилированию и активации ацетил-SКоА-карбоксилазы. Одновременно в клетке дефосфорилируется и инактивируется ТАГ-липаза.
Глюкагон, адреналин или другие гормоны, действуя по аденилатциклазному механизму с участием цАМФ-зависимой протеинкиназы, вызывают фосфорилирование и ингибирование ацетил-SКоА-карбоксилазы и, следовательно, останавливают липогенез. Одновременно они активируют ТАГ-липазу.
При уменьшении количества инсулина и возрастании глюкагона усиливаются липолиз в жировой ткани, поступление жирных кислот в печень и другие ткани и реакции их β-окисления. Такое состояние наблюдается при гипогликемии любого происхождения. При обратном соотношении гормонов начинаются реакции синтеза жиров.
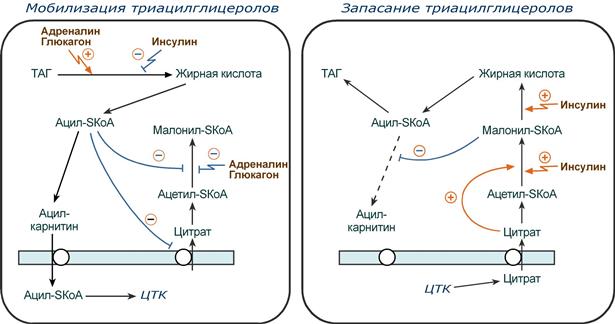
Способы регуляции реакций обмена триацилглицеролов
Метаболическая регуляция
В регуляции синтеза и окисления жирных кислот играют роль три участка:
1. Активность ацетил-SКоА-карбоксилазы регулируется:
- цитратом – аллостерический активатор фермента, накапливается в цитозоле при избыточном количестве энергии,
- ацил-SКоА по принципу обратной отрицательной связи ингибирует фермент, препятствуя синтезу малонил-SКоА. Т.е. если ацил-SКоА не успевает вступить в этерификацию или усиливается липолиз в клетке или увеличивается поступление жирных кислот извне, то автоматически блокируется синтез новых жирных кислот.
2. Транспорт цитрата из митохондрии в цитозоль подавляется избытком ацил-SКоА, это снижает синтез жирных кислот.
3. Карнитин-ацилтрансфераза ингибируется при образовании малонил-SКоА, что останавливает поступление ацил-SКоА внутрь митохондрий и снижает β-окисление.
Таким образом, когда в клетке имеется избыток энергии, то усиление синтеза жирных кислот достигается поступлением в цитозоль цитрата и при наличии малонил-S-КоА. Полученные молекулы ацил-SКоА быстро поступают на этерификацию глицерола до ТАГ и не накапливаются в цитозоле.
Если в клетке недостаточно энергии, то необходимо активировать β-окисление жирных кислот для ее получения. В этом случае гормональные влияния вызывают липолиз (или поступление жирных кислот извне) и накопление ацил-SКоА в цитозоле, что автоматически (см пункт 2) через уменьшение количества цитрата и малонил-SКоА препятствует синтезу новых жирных кислот. Параллельно (см пункт 3) убыль малонил-SКоА и активация карнитин-ацил-трансферазы запускает β-окисление.