Генетические заболевания, при которых происходит неполное расщепление полимерных веществ и их накопление, получили название лизосомные болезни накопления, так как они обусловлены дефектами специфических лизосомальных гидролаз. В случае накопления липидов такие болезни называются липидозы. При липидозах нарушается нормальный катаболизм липидов до соответствующих мономеров.
Липидозы
Болезнь Вольмана – редкое аутосомно-рецессивное заболевание из-за дефекта кислой эстеразы лизосом, что обусловливает накопление эфиров холестерола в лизосомах печени, селезенки, надпочечников, костного мозга и тонкого кишечника. Проявляется в первые недели жизни рвотой, диареей и стеатореей, гепатоспленомегалией и двусторонним кальцинозом надпочечников. Больные умирают в возрасте до 6 мес.
Болезнь Шюллера-Кристиана, аутосомно-рецессивное заболевание, характеризуется отложением в плоских костях, твердой мозговой оболочке и коже холестерола и его эфиров. Характерными являются деструктивные изменения в костях, остеопороз, мозжечковые расстройства. Заболевание обычно начинается в возрасте до 10 лет, реже позднее. Мужчины болеют в 2 раза чаще, чем женщины. Течение заболевания прогрессирующее. Дефектный фермент неизвестен.
Болезнь Гоше – отложение цереброзидов в макрофагальных клетках селезенки, печени, лимфатических узлов и костного мозга. Возникает в связи с аутосомно-рецессивным отсутствием гликоцереброзидазы (β-глюкозидазы). Основными симптомами заболевания являются спленомегалия, увеличение печени и селезенки, а также изменения в костях, проявляющиеся в виде остеопороза.

Дефектный фермент при болезни Гоше
При болезни Нимана-Пика наблюдается отложение сфингомиелина в клетках различных органов из-за дефицита сфингомиелиназы. Болезнь наследуется аутосомно-рецессивно, проявляется резким увеличением печени и селезенки, замедлением психического развития ребенка, появлением слепоты и глухоты. Чаще всего дети погибают в возрасте до 2 лет.

Дефектный фермент при болезни Нимана-Пика
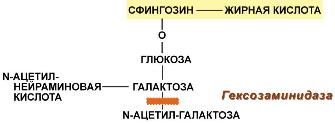
Болезнь Тея-Сакса (амавротическая семейная идиотия) является результатом дефекта N-ацетилгексозаминидазы, при котором происходит отложение ганглиозидов в клетках головного мозга, что сопровождается атрофией зрительных нервов, слепотой, слабоумием и смертью в младенческом возрасте.