Гликогеновые болезни – это наследственные заболевания, обусловленные недостаточностью каких-либо ферментов, отвечающих за метаболизм гликогена. Могут быть нарушены обе стороны обмена: как синтез гликогена, так и его распад. Средняя частота встречаемости составляет 1:40000.
Гликогенозы
Синдром гликогеноза возникает в результате дефекта фермента синтеза или мобилизации гликогена, что приводит к накоплению или изменению структуры гликогена в разных тканях, чаще в печени и мышцах. Следует отметить, что при гликогенозах количество гликогена не всегда изменено, изменения могут быть только в структуре его молекулы.
Всего известно 12 типов гликогенозов. По патогенетическому признаку гликогенозы делят:
- печеночные – 0, I, III, IV, VI, VIII, IX, Х, ХI типов,
- мышечные – V и VII типов,
- смешанные – II типа.
Печеночные гликогенозы
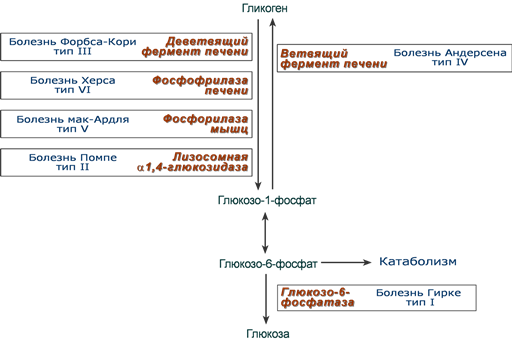
Самый частый гликогеноз I типа или болезнь фон Гирке (частота 1 : 50000-100000 новорожденных) обусловлен аутосомно-рецессивным дефектом глюкозо-6-фосфатазы. Из-за того, что этот фермент есть только в печени и почках, преимущественно страдают эти органы, и болезнь носит еще одно название – гепаторенальный гликогеноз. Даже у новорожденных детей наблюдаются гепатомегалия и нефромегалия, обусловленные накоплением гликогена не только в цитоплазме, но и в ядрах клеток. Кроме этого, активируется синтез липидов с возникновением стеатоза печени. Так как фермент необходим для дефосфорилирования глюкозо-6-фосфата с последующим выходом глюкозы в кровь, у больных отмечается гипогликемия и, как следствие, ацетонемия, метаболический ацидоз, ацетонурия.
Гликогеноз III типа или болезнь Форбса-Кори или лимит-декстриноз – это аутосомно-рецессивный дефект амило-α1,6-глюкозидазы, "деветвящего" фермента, гидролизующего α1,6-гликозидную связь. Болезнь имеет более доброкачественное течение, и частота ее составляет примерно 25% от всех гликогенозов. Для больных характерна гепатомегалия, умеренная задержка физического развития, в подростковом возрасте возможна небольшая миопатия.
При гликогенозе IV типа (болезнь Андерсена, 1% всех гликогенозов), связанного с дефектом ветвящего фермента, образуется гликоген с малым количеством ветвлений, что резко уменьшает скорость гликогенолиза.
Гликогеноз VI типа (болезнь Херса, 25% всех гликогенозов), связан с дефицитом печеночной фосфорилазы гликогена. При этом отсутствует мобилизация гликогена, развивается гепатомегалия и гипогликемия.
Мышечные гликогенозы
Для этой группы гликогенозов характерны изменения ферментов мышечной ткани. Это приводит к нарушению энергообеспечения мышц при физической нагрузке, к болям в мышцах, судорогам.
Гликогеноз V типа (болезнь Мак-Ардля) – отсутствие мышечной фосфорилазы. При тяжелой мышечной нагрузке возникают судороги, миоглобинурия, хотя легкая работа не вызывает каких-либо проблем.
Схематичное расположение дефектных ферментов при различных гликогенозах
Смешанные гликогенозы
Эти заболевания касаются и печени, и мышц, и других органов.
Гликогеноз II типа (болезнь Помпе, 10% всех гликогенозов) – поражаются все гликогенсодержащие клетки из-за отсутствия лизосомальной (кислой) α-1,4-глюкозидазы, поэтому данная болезнь относится к лизосомным болезням накопления. Происходит накопление гликогена в лизосомах и в цитоплазме. Заболевание составляет почти 10% всех гликогенозов и является наиболее злокачественным. Больные при отсутствии лечения умирают в раннем возрасте из-за кардиомегалии и тяжелой сердечной недостаточности.
Агликогенозы
Агликогенозы – состояния, связанные с отсутствием гликогена. В качестве примера агликогеноза можно привести наследственный аутосомно-рецессивный дефицит гликоген-синтазы. Симптомами является резкая гипогликемия натощак, особенно утром, появляется рвота, судороги, потеря сознания. В результате гипогликемии наблюдается задержка психомоторного развития, умственная отсталость. Болезнь несмертельна при адекватном лечении (частое кормление), хотя и опасна.